Systems Biology of Drug Perturbations
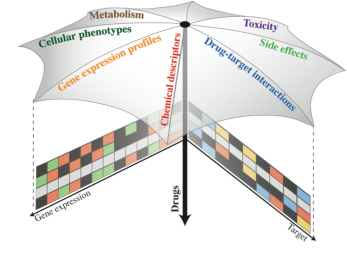 Combining and integrating different types of data (e.g.,
molecular in vitro data, data from cell-based assays, complex
phenotypic data such as side effects), we aim at gaining a better
understanding of drug mechanisms of action. Ideally, this will aid the
interpretation of drug action in many contexts, from the molecular
level (for instance, drug-target interaction) to the whole organism
(e.g., establishing links between side effects and cellular pathways:
Brouwers et al. PLoS
One, 2011; see also Iskar et al. Curr.
Opin. Biotechnol., 2012). To this end, we are developing methods
to predict protein targets, response pathways or side effects of drugs
from complex cell-based assays or organism-scale data and analyze
these predictive models to reveal the underlying biological
mechanisms. We see this as rational approaches to drug repositioning
and in silico drug safety assessment.
Combining and integrating different types of data (e.g.,
molecular in vitro data, data from cell-based assays, complex
phenotypic data such as side effects), we aim at gaining a better
understanding of drug mechanisms of action. Ideally, this will aid the
interpretation of drug action in many contexts, from the molecular
level (for instance, drug-target interaction) to the whole organism
(e.g., establishing links between side effects and cellular pathways:
Brouwers et al. PLoS
One, 2011; see also Iskar et al. Curr.
Opin. Biotechnol., 2012). To this end, we are developing methods
to predict protein targets, response pathways or side effects of drugs
from complex cell-based assays or organism-scale data and analyze
these predictive models to reveal the underlying biological
mechanisms. We see this as rational approaches to drug repositioning
and in silico drug safety assessment.
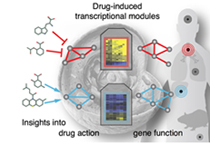 Currently, we focus on cellular assays of expression
changes upon chemical perturbations: The Connectivity Map
records gene expression (for >10,000 genes) in several cell lines for
hundreds of small molecules. Analysis of such complex multi-parametric
read-outs requires intelligent supervised (e.g. feature selection) and
unsupervised techniques (such as clustering and bi-clustering) in
order to reveal new drug mechanisms of action as well as functions of
the biological systems that respond to these treatments (see Iskar et al. Mol. Syst.
Biol., 2013).
Currently, we focus on cellular assays of expression
changes upon chemical perturbations: The Connectivity Map
records gene expression (for >10,000 genes) in several cell lines for
hundreds of small molecules. Analysis of such complex multi-parametric
read-outs requires intelligent supervised (e.g. feature selection) and
unsupervised techniques (such as clustering and bi-clustering) in
order to reveal new drug mechanisms of action as well as functions of
the biological systems that respond to these treatments (see Iskar et al. Mol. Syst.
Biol., 2013).
SIDER resource at EMBL | STITCH resource at EMBL | Drug modules at EMBL | CMap project at the BROAD
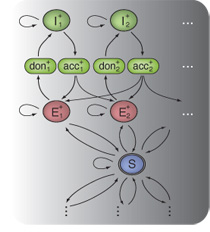 Together with colleagues from the Gunnar Rätsch's
group at the FML, I have been working on a Hidden Markov SVM-based
method called mTIM for transcript reconstruction from RNA-seq read
alignments. We obtained promising initial results for the
participation in the
Together with colleagues from the Gunnar Rätsch's
group at the FML, I have been working on a Hidden Markov SVM-based
method called mTIM for transcript reconstruction from RNA-seq read
alignments. We obtained promising initial results for the
participation in the 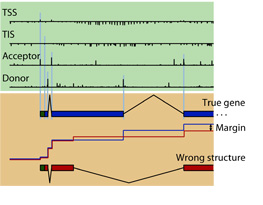 In Gunnar Rätsch's group we developed a novel and very accurate
gene finding system called mGene, which uses the latest advances in
machine learning, namely a discriminative structure prediction
technique called hidden semi-Markov SVMs (
In Gunnar Rätsch's group we developed a novel and very accurate
gene finding system called mGene, which uses the latest advances in
machine learning, namely a discriminative structure prediction
technique called hidden semi-Markov SVMs (