
Program Overview
Input
Sircah takes as input transcript models in the GFF3 format allowing the user the flexibility to choose the sources of evidence for the use in detecting alternative transcription. Such transcript models may come from the gene prediction pipelines of genome databases or from spliced alignments of ESTs or proteins against the genome. Within the GFF3 file the user may specify the completeness of the transcript model by setting the Bounded attribute in the GFF file. The attribute can have the following values: 1 (a complete transcript), 0 (an incomplete transcript), 3 (the transcript is complete at the 3' end) or 5 (the transcript is complete at the 5' end). If nothing is specified then EST_match types are considered incomplete and mRNA and cDNA_match types are considered complete. Additionally the user may use the Tags attribute to specify a comma-separated list of labels that can be used to analyse the alternative transcripts in subsets of the data. See the tutorial for details.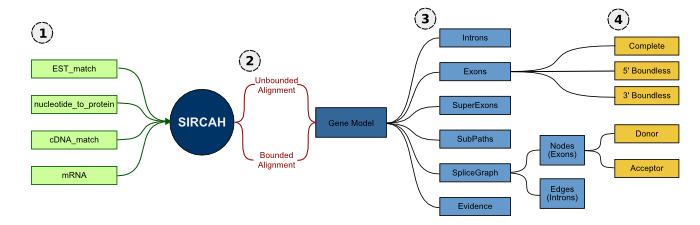
Sircah data models. (1) Transcript models are provided to Sircah in GFF3 format. (2) Sircah treats these alignments as either unbounded or bounded depending on whether they come from full length transcript models or not. (3) The transcript models are then used to create a gene model consisting of exons, introns, superexons (clusters of overlapping exons), a splice graph, subpaths (transcripts) and evidence (a mapping of the input data to subpaths). (4) Based on whether they're enclosed by introns or from a full-length transcript exons are further classified as boundless or complete, and based on their position within the splice graph they're classified as donors or acceptors.
Detection of Alternative Splicing and Transcription Events
Sircah uses a splice graph data model as to represent the transcripts models in a non-redundant form. The nodes of the directed graph are exons, the edges introns and transcripts are represented as subpaths of the graph. Additionally overlapping exons are clustered into superexons. A series of rules are then applied to the data and based on the topology of the splice graph and the membership of the superexons the following alternative events can be classified: alternative initiation exons, alternative termination exons, exons with alternative 3' and/or 5' splice sites, retained introns and skipped exons.
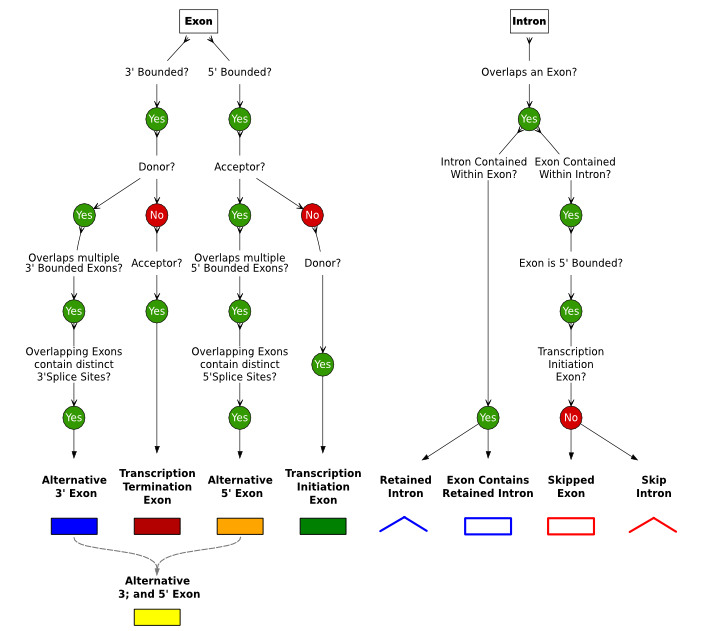
Rules used by Sircah to detect alternative splicing. Based on the data model described in INSERT REFERENCE alternative initiation exons, alternative termination exons, exons with alternative 3' and/or 5' splice sites, retained introns and skipped exons are classified.}
Visualisation of Alternative Splicing and Transcription Events
The splice graph and the transcript models used to construct it can be visualised in a variety of ways to show: (i) the alternative events detectable and the transcript models used, (ii) the different events detectable in subsets of the data and (iii) the coverage of introns and exons by transcript models. The visualisation is created in the SVG format, allowing the creation of publication quality images. In addition the ids of all the elements in the SVG file are directly mappable to the ids used in the data objects, allowing the user to alter graphic after it's generated and even create interactive graphics using javascript.
Analysis of Subsets of Evidence
One of the most powerful features of Sircah is the ability to create splice graphs based on a subset of the total data. This allows the user to compare alternative transcription events under different conditions. For example by tagging the EST alignments with the EVOC expression ontology one can examine the tissue distribution of alternative transcription events.
Data Serialisation
In order to be able to carry out such analyses it is important to be able to save and reload the data models described above. To facilitate this Sircah can serialise its data objects to either an XML file or to a relational database using the SQLAlchemy python module.


